4 Introduction to R: Part I
If you are currently participating in a timetabled BIOS103 QS workshop, please ensure that you cover all of this section’s content and complete this week’s formative and summative assessments in the BIOS103 Canvas Course.
We are going to diverge from what you have been doing in the labs this week in order to focus on introducing you to R. Our learning objectives are as follows:
- Start a new R Project in R Studio
- Read a dataset to a variable
- Inspect a dataset
- Subset a dataset by slicing and filtering
- Create summary tables of descriptive statistics
- Sort a table
You should have completed section 1.2 already and have R and R Studio installed on your personal machine. If you are unable to install R and RStudio then please use one of the University computers.
OMG. Why, are you making me learn to code?
In my experience, at least half of you will love learning to code. The rest of you will hate my guts. I’m OK with that. That’s because I truly believe that in the near future you will see that even a basic understanding of coding gives you a huge advantage in your studies and future careers. However, if you really need me to give you explicit reasons as to why I’m making you learn to code then here are six:
1. Data Literacy
All the biosciences rely heavily on data analysis. Learning to code will equip you the skills to efficiently manipulate and interpret complex datasets. R, in particular, is well-suited for handling bioscientific data, from genomic sequences to clinical trials and environmental modelling.
2. Reproducibility
Coding promotes transparency and reproducibility in scientific analyses. Unlike manual methods, where calculations or procedures may be difficult to replicate, scripts provide a clear, step-by-step record that can be easily shared and re-run. This is vital for ensuring the integrity of scientific research.
3. Automation
Bioscientists often work with large datasets, and coding enables the automation of repetitive tasks such as data cleaning, statistical analyses, and reporting. This not only saves time but also minimises human error, allowing students and researchers to focus on interpreting their results.
4. Career Readiness
Coding has become an indispensable skill in the biosciences. Whether working in research, biotechnology, or environmental consultancy, understanding how to work with data in R (or similar tools) will give you a competitive edge when applying for jobs or pursuing further academic research.
5. Critical Thinking
Writing code fosters critical thinking and problem-solving skills. While learning to code you will have to break down complex problems, troubleshoot errors, and develop logical workflows. These skills are not only useful for coding but are also vital for scientific thinking and research.
6. Integration with Lab Skills
Coding complements traditional lab skills. In the era of bioinformatics and systems biology, being able to analyse experimental data computationally can provide additional insights that are often not apparent from lab experiments alone.
Have you accepted your fate then? Good. Now lets get something else straight: I am NOT going to teach you to code. You are going to do that yourself, over many months (and probably years). This book represents a mere “dipping of the toes” into the world of R coding and is by no means comprehensive. In fact, it is completely and utterly incomplete. You will need to fill in many of the blanks yourself as you encounter them. How you go about this is up to you, but checkout the Appendix for good places to start.
4.1 Reading and Inspecting Data
Let’s dive in then! This week we’re going to be introducing you to some fundamental data handling concepts in R using the fantastic Pantheria dataset of extant and recently extinct mammals.
Open RStudio and start a new project
Download the data from here (or right click, save as from here).
Copy the downloaded file into your RStudio project directory (folder). You should see the file appear in your Files window in the bottom-right corner of RStudio.
Create a new R script file and save it with a sensible filename (e.g. pantheria_summary.R)
Read the data to a variable called
pantheria_data: In your new script file, write the following line of code on line 1:
Run the line by clicking anywhere on the line and then clicking the run button (in the top-right corner of your script window) or by pressing Ctrl+Enter on your keyboard. You should see a new line appear in your Environment window in the top-right of your RStudio like in figure 4.1. If you don’t see it then the here are the most common reasons as to why:
- You haven’t used quotation marks around your data filename
- You’ve mis-spelled your filename
- You haven’t put your data file in the correct folder
- You didn’t create an R project first.
Figure 4.1: Details of your data variable should appear in your environment window after running line 1.
Figure 4.2: By clicking on your data variable in your environment window you can inspect the data in RStudio
What is a variable in R?
A variable in R is a symbolic name representing a value stored in memory. Variables are created by assigning values using
<-.Naming Convention
I prefer using lowercase words for variable names. If multiple words are needed, I separate them with an underscore “_“, avoiding spaces. For example: my_variable.
Best Practices
- Descriptive Names: Choose clear, meaningful names.
- Consistency: Use the same naming style throughout your code.
- Avoid Reserved Words: Don’t use R’s reserved keywords (
if,else,for, etc.)- Uniqueness: Ensure variable names are unique within your environment.
These practices enhance code readability and maintainability.
Well done if you can see your data variable! You can click on your variable (in the environment window) and inspect your data directly. You should see something like that shown in figure 4.2. Wow! Look at all that data. Yummy. Now the world is your oyster, as they say. From here you can go in any number of directions and use R to summarise, visualise and analyse your data in order to gain novel insights.
Let’s start small by asking the very simple question:
“What order of mammal occurs the most frequently (has the most number of rows) in the dataset?”.
To answer this question, I need to go through my data row by row and count how many times a row corresponding to each order (e.g. Dermoptera, Chiroptera, Rodentia etc …) appears. Let’s extend my R script as follows:
Let’s break these additional lines down, step-by-step:
Firstly, you’ll notice that I’ve left lines 2 and 5 blank. You don’t need to do this, but I like to because it gives my code a bit of breathing space and is often easier to identify a problem later.
In line 3, I’m using the
table()function to count the number of times a unique value in my order column appears. By puttingdata$Orderinside the table function’s brackets we are “passing” it all of the values in the Order column as a big list. I’m then assigning the output of the table function to a new variable calledordersso that I can refer to it later.Lastly in line 5, I’m using a print function to output the contents of the variable
ordersto my console as shown in figure 4.3
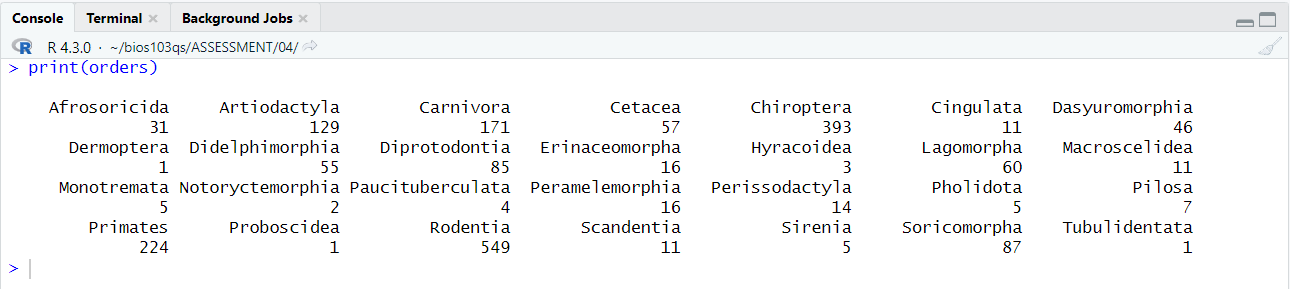
Figure 4.3: When using print(orders) the following should appear in my RStudio console.
What is a function in R?
A function in R is a set of instructions that performs a specific task or calculation, taking inputs (arguments) and returning an output (result). So far in this course, we have used three pre-defined functions: read.csv() to read data from a CSV file, table() to summarise categorical data, and print() to display output in the console. While these functions are built into R, we can also create our own functions when we need to perform customised tasks—more on this later in the course.
You can see by inspecting your console output that the order Rodentia is the most frequently occurring order with 549 rows in the data.
But what if there were lots more orders? It might not be so straightforward. Let’s get our R script to pick out the most frequently occurring order for us:
pantheria_data <- read.csv("pantheria_999.csv")
orders <- table(data$Order)
most_common_order <- names(which.max(orders))
print(most_common_order)I’ve inserted the line most_common_order <- names(which.max(orders)) at line 5. Let’s break it down:
which.max(orders):
- Function:
which.max()is a built-in R function that returns the index of the first maximum value in its input. - Input: In this case, the input is
orders, which is the table we created earlier containing the counts of each order in the dataset. - Output: The function identifies the index position of the maximum count (i.e., the highest frequency) in the
orderstable. In our case “Rodentia” is at position 24 in the table.
names(...):
- Function: The
names()function retrieves the names (or labels) associated with the elements of an object. For example if I usednames(orders)the function would simply return a list of all the unique orders. - Usage: By wrapping
which.max(orders)insidenames(), we are saying, “give me the name of the order that corresponds to the maximum frequency index returned bywhich.max().” - Output: In our case the name “Rodentia” is returned.
most_common_order: I’ve assigned the result of mynames(which.max(orders))to a new variable using<-.Finally, I’ve updated my print statement to `print(most_common_order)’. This should now print out “Rodentia” in my console when I re-run my script.
Phew! That took a bit of explaining. Make sure you understand what just happened. The wrapping of a function in a function is a common practice in R and things can quickly become obfuscated. It’s good practice to comment your code so that you (or someone else) can quickly make sense of what is going on. Here is my final code, with comments!
# Read the CSV file and store its contents in 'data'.
data <- read.csv("pantheria_999.csv")
# Create a frequency table of the 'Order' column.
orders <- table(data$Order)
# Get the name of the most common order.
most_common_order <- names(which.max(orders))
# Print the most common order to the console.
print(most_common_order)4.2 Subsetting
In data analysis, subsetting refers to the process of extracting a portion of a dataset based on specific criteria. This allows you to focus on the most relevant information, making analysis more efficient and tailored to your needs. There are two key methods for subsetting data in R: Slicing and Filtering.
4.2.1 Slicing
Slicing is a crucial operation in data analysis, enabling you to extract specific rows, columns or both from an existing data frame. This technique helps streamline workflows, especially when working with large datasets, by narrowing down data to focus on relevant sections.
Slicing Example
Let’s add a new line to our earlier script file:
This code creates a new variable called slice and assigns to it the first 1,000 rows and the first 10 columns of the dataset data.
Run the line and you should see a new dataframe called slice appear in your Environment window (top-right).
What if I’d wanted to select ALL the rows but still slice off the first 10 columns? Well, that would look like this:
And what if I don’t want to use index values to select the columns (i.e., 1:10) and I want to select them by name? For example let’s say I’ve inspected the column headers and I specifically want to select the following columns:
- Order
- AdultBodyMass_g
- BasalMetRate_mLO2hr
I can do that using this code:
Here I’ve defined a vector using the c() function containing the verbatim names of columns I want and used this instead of specifying a range of column indices.
4.2.2 Filtering
Unlike slicing, which selects a subset of rows and columns based on their position, filtering involves evaluating each row against a set of logical conditions and including only those that meet the criteria.
Filtering Example
In the above slicing example we created a dataframe variable called slice which contains three columns. If you click on the variable in the environment window to select it you’ll see something like that shown in 4.4.
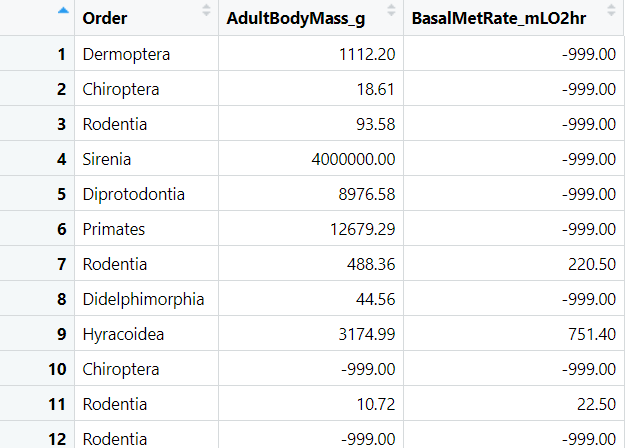
Figure 4.4: You’ve sliced your data but you’ll need to be able to filter in order to remove the -999 values.
Hmmm, that’s weird. What’s with all the -999 values? In this dataset the value -999 has been used to indicate where a value wasn’t measured. I can remove these -999 values in my slice dataframe like this:
Let’s break this down:
filtered <-:
- This assigns the result of the filtering operation to a new variable called filtered. This variable will contain only the rows from the slice data frame that meet certain conditions.
slice[slice$AdultBodyMass_g >= 0 & slice$BasalMetRate_mLO2hr >= 0, ]:
- This part performs the actual filtering operation on the slice data frame.
slice$AdultBodyMass_g:- This accesses the AdultBodyMass_g column of the slice data frame. The $ operator is used to refer to a specific column.
slice$BasalMetRate_mLO2hr:- Similarly, this accesses the BasalMetRate_mLO2hr column of the slice data frame.
slice$AdultBodyMass_g >= 0:- This condition checks each value in the AdultBodyMass_g column to see if it is greater than or equal to 0, generating a logical vector (TRUE or FALSE) for each row.
slice$BasalMetRate_mLO2hr >= 0:- This condition checks each value in the BasalMetRate_mLO2hr column to see if it is greater than or equal to 0, producing another logical vector.
&:- This is the logical AND operator, which combines the two logical vectors created by the previous conditions. The result is a new logical vector that is TRUE only for rows where both conditions are TRUE.
- slice[… , ]:
- The entire expression inside the square brackets is used to subset the slice data frame. The rows for which the combined condition is TRUE are selected, and all columns (indicated by the empty space after the comma) are returned.
In summary, the code creates a new data frame, filtered, which contains only the rows from the slice data frame where both the AdultBodyMass_g and BasalMetRate_mLO2hr values are greater than or equal to 0.
4.2.3 So what?
Well, being as you’ve gone to all that trouble to slice and filter your data we should probably do something spectacular with it! Try adding the following code to your script to and running it.
plot(slice$AdultBodyMass_g, slice$BasalMetRate_mLO2hr,
log="xy",
xlab = "Adult Body Mass (g)",
ylab = "Basal Metabolic Rate (mLO2/hr)",
main = "")You should see something that looks like what is shown in figure 4.5.
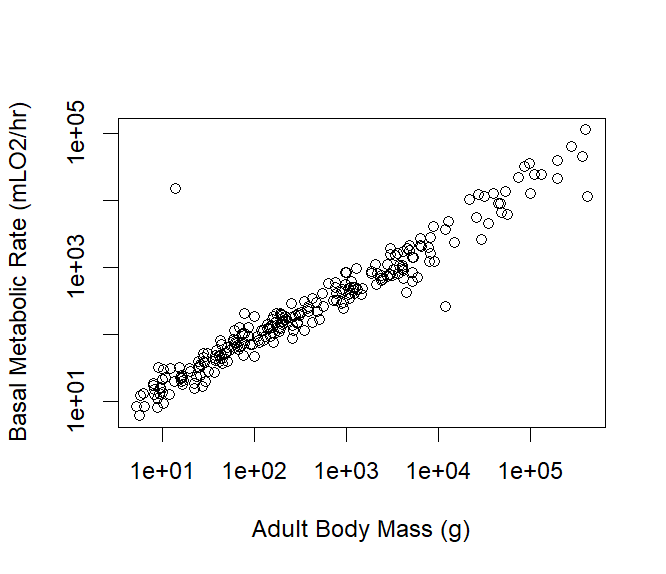
Figure 4.5: An all species log-log scatterplot of mammal adult body mass (g) vs basal metabolic rate (mLO\(_2\)/hr)
Notice that I’ve used the argument log="xy" within my plot() function to make both the x and y axes scale logarithmically. This means that instead of the axes increasing in equal increments (e.g., 1, 2, 3), they increase by factors of ten (e.g., 10, 100, 1000).
This is particularly useful when the relationship between two variables spans several orders of magnitude, as is often the case in biological data. When both axes are scaled logarithmically, relationships that might look curved on a linear scale can appear as straight lines, making it easier to identify patterns.
In this case, the plot reveals a linear relationship between the two variables (e.g., adult body mass and basal metabolic rate) when viewed on a log-log scale. This linearity indicates that as one variable increases by a certain percentage, the other variable increases by a consistent percentage, rather than by a fixed amount. This relationship is described as allometric scaling and is remarkably common in nature.
What’s fascinating is that this log-log linearity holds across all mammals! From the smallest mouse to the largest whale, body mass and metabolic rate follow a consistent scaling relationship when plotted logarithmically. This insight allows scientists to understand fundamental principles about how biological processes like metabolism are related to size in the animal kingdom.
4.3 Summarising Data in R
Remember how in chapter 2 we generated a summary table of descriptive statistics using Excel? We can do it in R too!
As an example, let’s answer the following question:
What are the top 10 families with the highest number of neonate body mass observations, along with their summary statistics (mean, median, max, min, and standard deviation)?
The following script will do the job, as you can see by the accompanying output.
# Read the CSV and filter out invalid rows
data <- read.csv("pantheria_999.csv")
data_filtered <- data[data$NeonateBodyMass_g != -999, ]
# Calculate summary statistics for each Family
summary_table <- aggregate(NeonateBodyMass_g ~ Family, data_filtered, function(x)
c(Mean = mean(x), Median = median(x), Max = max(x), Min = min(x), SD = sd(x), Count = length(x)))
# Convert the list columns to individual columns
summary_table <- do.call(data.frame, summary_table)
# Rename the columns
colnames(summary_table) <- c("Family", "Mean", "Median", "Max", "Min", "Std. Dev.", "Count")
# Format all numeric values to 1 decimal place (except Count)
summary_table[, 2:6] <- round(summary_table[, 2:6], 1)
# Sort by Count in decreasing order and return top 10 most frequently occurring families in data
summary_table <- summary_table[order(-summary_table$Count), ][1:10, ]
# Write to CSV and print
write.csv(summary_table, "summary_table.csv", row.names = FALSE)
print(summary_table)## Family Mean Median Max Min Std. Dev. Count
## 9 Bovidae 8291.2 4819.6 39843.1 500.0 8521.8 36
## 17 Cercopithecidae 465.9 450.0 890.0 240.7 156.7 19
## 69 Mustelidae 149.7 30.0 1894.4 2.0 429.0 19
## 18 Cervidae 4252.0 3082.2 13500.0 400.0 4027.9 18
## 13 Canidae 167.6 102.1 412.3 28.0 127.6 14
## 47 Heteromyidae 3.1 3.0 7.7 1.0 1.9 14
## 80 Phocidae 16490.5 15122.7 39393.0 3050.0 10434.1 14
## 93 Sciuridae 10.3 6.1 33.0 3.3 10.0 13
## 41 Felidae 186.4 161.6 409.9 72.0 102.4 12
## 56 Leporidae 74.4 90.0 123.0 25.9 39.0 11See if you can reproduce the output by creating a new script file in your project, copying and pasting the code above and clicking the Source button in the top right of your script editor window.
What Next?
I don’t need you to understand every line in the script. In fact there are some lines (6 -7 in particular) that are doing some seriously funky stuff that took me years to learn and understand fully. Nonetheless, ask yourselves what would you need to tweak to get the code to generate a summary table for another variable (e.g. AdultBodyMass_g) or group the results by Order instead of Family.
Often, when learning to code, it can feel overwhelming trying to understand every single detail of a script before running it. However, there’s value in a “run it and see what happens” approach, especially when you’re starting out. The beauty of coding is that you don’t need to fully grasp every element in order to get results.
In fact, many experienced coders begin with scripts they might not completely understand. The key is knowing just enough to use the script to answer your question. Once you see the output and observe how the code works, you can begin to tweak and modify it to suit a different dataset or to refine your results.
The process of experimenting with the code helps deepen your understanding over time. You’ll gradually learn how each part of the script contributes to the output, and that’s where real learning happens.
Turbo charge your learning with Chat-GPT
I have serious mis-givings about asking generative AI to generate code out of thin air. In my experience, GAI likes to show off and often over-complicate things. This can seriously confuse students who are just starting to learn to code.
However, using it to understand existing code? Yes! Do it! What a fantastic way to learn. I use it every day when it comes to code.
I cut and pasted the script above into Chat-GPT and gave it the following prompt: “Explain in detail”.
You can see the result here. Isn’t that amazing?
Feedback Please.
I really value your feedback on these materials for quantitative skills. Please rate them below and then leave some feedback. It’s completely anonymous and will help me improve things if necessary. Say what you like, I have a thick skin - but feel free to leave positive comments as well as negative ones. Thank you.
4.4 Complete your Weekly Assignments
In the BIOS103 Canvas course you will find this week’s formative and summative QS assignments. You should aim to complete both of these before the end of the online workshop that corresponds to this section’s content. The assignments are identical in all but the following details:
- You can attempt the formative assignment as many times as you like. It will not contribute to your overall score for this course. You will receive immediate feedback after submitting formative assignments. Make sure you practice this assignment until you’re confident that you can get the correct answer on your own.
- You can attempt the summative assignment only once. It will be identical to the formative assignment but will use different values and datasets. This assignment will contribute to your overall score for this course. Failure to complete a summative test before the stated deadline will result in a zero score. You will not receive immediate feedback after submitting summative assignments. Typically, your scores will be posted within 7 days.
In ALL cases, when you click the button to “begin” a test you will have two hours to complete and submit the questions. If the test times out it will automatically submit.